
🏏ICC Men's T20 World Cup 2026: First Semi-Final- SA vs NZ🏏

HOLIKA DAHAN 3.3
I Love You Armaan

Toxic Postponed To 4th June 2026
Abhimaan Ki 4th Shaadi
With is with those expressions? Alia's interview at Bafta
HOLI GADBAD 4.3
Why was Dharundar snubbed at Zee Awards
Abeekmaan are cringe
Happy birthday to Shraddha Kapoor

Avatar & Signatures Shop BC 2026 | V-Day Celebration | VOTING ON!

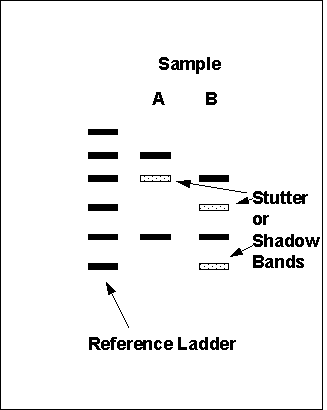
| DNA Examinations | ||||||
| ||||||
|
There are two sources of DNA used in forensic analyses. Nuclear DNA (nDNA) is typically analyzed in evidence containing blood, semen, saliva, body tissues, and hairs that have tissue at their root ends. Mitochondrial DNA (mtDNA) is typically analyzed in evidence containing naturally shed hairs, hair fragments, bones, and teeth. | ||||||
| Documenting, Collecting, Packaging, and Preserving DNA Evidence If DNA evidence is not properly documented, collected, packaged, and preserved, it will not meet the legal and scientific requirements for admissibility in a court of law.
When DNA evidence is transferred by direct or secondary (indirect) means, it remains on surfaces by absorption or adherence. In general, liquid biological evidence is absorbed into surfaces, and solid biological evidence adheres to surfaces. Collecting, packaging, and preserving DNA evidence depends on the liquid or solid state and the condition of the evidence. Blood Examinations Collecting Known Samples Blood | ||||||
Only qualified medical personnel should collect blood samples from a person.
Identify each tube with the date, time, subject's name, location, collector's name, case number, and evidence number.
Pack liquid blood tubes individually in Styrofoam or cylindrical tubes with absorbent material surrounding the tubes. Label the outer container KEEP IN A COOL DRY PLACE, REFRIGERATE ON ARRIVAL, and BIOHAZARD. | ||||||
Blood on a Person | ||||||
| ||||||
Blood on Surfaces or in Snow or Water | ||||||
| ||||||
|
| ||||||
Air-dry small suspected wet bloodstained objects and submit the objects to the Laboratory. Preserve bloodstain patterns. Avoid creating additional stain patterns during drying and packaging. Pack to prevent stain removal by abrasive action during shipping. Pack in clean paper. Do not use plastic containers. When possible, cut a large sample of suspected bloodstains from immovable objects with a clean, sharp instrument. Collect an unstained control sample. Pack to prevent stain removal by abrasive action during shipping. Pack in clean paper. Do not use plastic containers. | ||||||
|
Claims made by the suspect(s) regarding the source of the blood. Whether animal blood is present. Whether the stains were laundered or diluted with other body fluids. | ||||||
Semen and Semen Stains | ||||||
Submit small suspected dry semen-stained objects to the Laboratory. Pack to prevent stain removal by abrasive action during shipping. Pack in clean paper. Do not use plastic containers. When possible, cut a large sample of suspected semen stains from immovable objects with a clean, sharp instrument. Collect an unstained control sample. Pack to prevent stain removal by abrasive action during shipping. Pack in clean paper. Do not use plastic containers. | ||||||
Seminal Evidence From Sexual Assault Victim(s) | ||||||
| ||||||
Buccal (oral) swabs | ||||||
Air-dry the swabs and place in clean paper or an envelope with sealed corners. Do not use plastic containers. Identify each sample with the date, time, subject's name, location, collector's name, case number, and evidence number. | ||||||
|
| ||||||
Submit suspected small, dry saliva- or urine-stained objects to the Laboratory. Pack to prevent stain removal by abrasive action during shipping. Pack in clean paper or an envelope with sealed corners. Do not use plastic containers.
Pick up chewing gum with gloved hands or clean forceps. Air-dry and place in clean paper or an envelope with sealed corners. Do not use plastic containers. | ||||||
Hair | ||||||
Tissues, Bones, and Teeth Call the Laboratory at 703-632-7572 prior to submitting suspected tissues, bones, or teeth to ensure that the evidence will be accepted for examination. The communication accompanying the evidence must reference the telephone conversation accepting the evidence.
nonrestored premolar. nonrestored canine. nonrestored front tooth. restored molar. restored premolar.
| ||||||
Much of what has been discussed previously is involved in crime scene processing, so there is little need to rehash. There remain a number of points that fit into the categories of trace evidence, the homicide victim and a few administrative matters to get the nitty gritty explanation of forensic science out the way and conclude this set of web pages. This is more the loose ends section than anything, a smattering of quick explanations for topics that don't appear in the other categories. But first, some review. Most physical evidence concerns itself with class characteristics and individual characteristics. Class characteristics are those characteristics which are common to a group of similar objects. For example, you buy a pair of Air Jordan sneakers. All Air Jordan sneakers have the same shape and same tread design on the bottom. These are class characteristics. Individual characteristics are those characteristics which are unique to a given object and set it apart from similar objects. You wear your Air Jordans around for awhile and they get worn. The treads wear down. They get little pits and gouges in them. These little pits and gouges are individual to your shoes and no others since no one has walked over the exact same surfaces in the exact same way in their Air Jordans. These concepts apply to all kinds of forensic evidence, from soil to glass, from rope to hefty bags. The fracture match is another important concept, particularly when trace evidence is concerned. Tear a piece of paper in half. Hold the two halves together. This is called a fracture match. No two tears are exactly alike. One half of a tear can always be matched back to its other half. Remember that. If a half of something found at a crime scene can be matched to the other half of something found on a suspect, that's damn good physical evidence. Crime Scene Documentation. The first thing you do after securing a crime scene is document it. Always take pictures. They are the best record available. They show the crime scene as it was found; where objects are in relation to other objects, victims, rooms, etc. Take notes. Describe the scene, its over all conditions. Describe rooms, lights, shades, locks, food; anything that can indicate a time frame, condition of scene or that might have even the slightest evidentiary significance. Check dates on mail and newspapers. Diagram the crime scene. Take measurements. Photos are good to show where an object is in relation to another object, but measurements tell exactly how far. Chain of Custody. Chain of Custody is of paramount importance to any investigation. It is the unbroken sequence of events that is caused by an item of evidence from the time it is found at the crime scene to the time it appears in court. Every link in this chain is documented, from discovery at the crime scene, through evidence gathering, storage, lab analysis, return to storage, transfer to court. Every link is documented by date, time, handling individual, what was done with the evidence by that individual. If chain of custody is broken, if the evidence cannot be accounted in one step of its journey from crime scene to court room, it is rendered inadmissible; useless to the case. Locard's Exchange Principal. Every time an individual comes in contact with a place or another individual, something of that individual is left behind at the place, and something of that place is taken away with the individual. If your Aunt Bertha gives you a big hug and walks away, fibers from her clothes will be on your clothes and fibers from your clothes will be on hers. Your hair is constantly falling out (circle of life, guys). You leave it all over the place. Just look around your house. You pick up carpet fibers on your shoes, dirt from the ground. Your skin flakes off. Look at the Wayne Williams case in Atlanta. He was convicted because fibers found on the body of one of his victims matched fibers from the carpet in his house. Might not sound like much, but it's GREAT physical evidence. Entomology. To paraphrase Indiana Jones, "Bugs. I hate bugs..." But they are great evidence and can be used to determine time of death. In some cases, they can be used to determine if a body has been moved from one geographic location to another. Certain bugs incubate and hatch at certain known rates. If bugs are found on a corpse, the age of the bugs can be extrapolated backward to estimate time of death. Blood Spatters. Blood spatters help a great deal in recontructing a crime scene. They can be used to corroborate or disprove and alibi. They can be used to convict the guilty. There is much more to it than looking at a stain or spatter and saying, "This is where the crime took place." The patterns of the spatters and the shapes of the individual blood droplets themselves can tell how the crime was committed. Drops falling from different heights (i.e. at different speeds) will leave different looking spatters. A drop falling from a low height of a few inches will leave a small cohesive circle. At greater heights, the circle will be larger and may even have a 'crown' effect. Hitting a surface at an angle does even more to disrupt a blood droplet. Perpendicular impact leaves a droplet fairly uniform, as shown below. A droplet hitting a surface at an angle will bulge out in one direction, indicating the direction of travel of the droplet. Cast off stains are a result of blunt force trauma (beating with an object such as a hammer). Pulling back from a blow produces a blood spatter that indicates direction, by creating an arc of blood droplets. You can determine the number of blows inflicted by counting the arcs. You can also determine the orientation of the individuals involved, the size of the object used and the right or left handedness of the assailant. Click here to go to the next page. |
| Glass Examinations Glass comparison examinations can determine whether particles of glass originated from a broken source of glass. Glass fracture examinations can determine the direction and type of the breaking force and the sequencing of shots. Questions concerning glass evidence should be directed to 703-632-7690. Follow the Evidence Submission directions including Requesting Evidence Examinations and Packaging and Shipping Evidence. Comparison
Submit the victim(s)' and suspect(s)' air-dried clothing. Each item must be packaged separately in a paper bag. Search for particles in the victim(s)' and suspect(s)' hair, skin, and wounds. Submit particles in leakproof containers such as film canisters or plastic pill bottles. Do not use paper or glass containers. Search for particles in vehicles by vacuuming each section of the vehicle separately. Do not use tape for recovering glass particles. Submit vacuum sweepings in leakproof containers. Do not use paper or glass containers. Ship known and questioned debris separately to avoid contamination. | |
Fracture
Submit all glass pieces so that the pieces can be fitted together to identify the radial cracks near and at the point(s) of impact and to increase the probability of matching edges. Pack all glass separately and securely to avoid shifting and breaking during shipping. Submit the entire piece of laminated glass, if possible. Secure the glass between plywood or sturdy cardboard. Do not place any objects into the impact area. | |
< name=forsbar> </> Forensic Photography. I'm not going to go into the basics of photography here. There are other pages on the web that will do it much better than I can. Actually, this section is going to be pretty short. There are just a few particulars to hit on, and a couple of interesting techniques. The first thing that needs to be done after securing the crime scene is photographing it. This creates a permanent record of the condition of the crime scene, one that is incontestable. First, take a picture that shows where the scene is; a shot with a street sign with the crime scene location in the background. Take pictures of the areas around the crime scene; alleys, dumpsters, rear areas, neighboring structures and even the structures across the street. Next, take pictures of the outside of the structure, showing points of entry and exit. Enter the structure, taking shots that show the locations and layout of the rooms. Take pictures of the whole room where the crime took place. Take close-ups of the scene or body. All pictures of items of evidence, which will be covered in the next paragraph, should be take both with and without a scale (a small ruler showing the size of the object). Take pictures with the scale to show the size of an object. Take pictures without the scale in case its presense in the picture blocks other evidence. What items are photographed at a crime scene? Bullet casings; photograph as a group and photograph individually. Photograph any dropped items, foot prints or animal tracks. If a homicide, photograph the body or bodies. Photograph any toolmarks, bitemarks or skin impressions. Basically, anything that might be evidence is photographed. Imprint evidence requires extra measures. Shoe imprints are photographed individually and as a series or group. Shoe imprints need to be lit from the side to show as much detail in the imprint as possible. Tire imprints are photographed from above as a whole. If the tire imprint is four feet long, then a picture showing all four feet is taken. Detail pictures are then taken showing one foot sections, each picture overlapping the one before it. This way, specific detail can be show and the overlapping pictures lined up to show the whole print. Again, all pictures are take with and without a scale. There is a special technique for no light situations. This technique is useful outdoors at night (perhaps a car accident scene), or in situations where the room is too big to light or there is no light available for pictures to be taken (such as a burnt out warehouse arson). The camera is set on a tripod with its shutter locked open. The photographer walks to several points in the room, popping off the flash, which is held in his or her hand. Each time the flash goes off, the film in the camera is exposed to another part of the room. The photographer does not appear as he/she is behind the flash and does not get exposed to the light when it pops off and only moves around the room while it is still dark. Remember, the film in a camera captures light. If there is no light, you can walk around in front of a camera all you want and never show up on the film. Video is also used to film crime scenes, taking long sweeping shots that take in everything in an unbroken time frame. The problem with video is, camcorder microphones will pick up the officers talking in the background, which can sometimes be embarrassing when the tape is replayed in court. | |
| Gunshot Residue on Hands Examinations For muzzle-to-target distance determinations involving gunshot residue, see Firearms Examinations. When a firearm is discharged, vaporous and particulate materials called gunshot residue (GSR) are expelled. After collecting gunshot residue from a suspected shooter's hands, the major elemental components of most cartridge primer mixtures can be analyzed to associate a suspect with the recent discharge of gunpowder from a firearm. This examination is used to determine if a person was in the presence of gunshot residue within a limited time period after a weapon discharge. The Laboratory provides gunshot residue examinations to assist FBI field office investigations only. Questions concerning gunshot residue evidence should be directed to 703-632-8441. Follow the Evidence Submission directions including Requesting Evidence Examinations and Packaging and Shipping Evidence.
Usually gunshot residue examinations will only be performed when samples are collected from living persons' hands. Gunshot residue evidence must be collected within five hours of exposure to the discharge of a firearm. Complete the information sheet included with the gunshot residue collection kit, and follow the instructions for collecting evidence. | |
| Hairs and Fibers Examinations Hairs | |
| Fibers Questions concerning hairs and fibers evidence should be directed to 703-632-8449. Follow the Evidence Submission directions including Requesting Evidence Examinations and Packaging and Shipping Evidence.
| |
| Image Analysis Examinations Photographic Comparisons Examinations of film, negatives, digital images, photographic prints, and video recordings including surveillance images involve comparisons of subject(s) depicted in the questioned images with known images (e.g., photographs, videos) of suspect(s). Similar comparisons can be made between the subject(s)' clothing and clothing seized from the suspect(s). Comparisons can also be made with firearms, vehicles, and other objects depicted in surveillance images. | |
| Photogrammetry Dimensions can be derived from photographic images through the use of geometric formulae or on-site comparison. Examples of photogrammetry include determining the height of bank robbery subject(s) and the length of the weapon(s) used by the subject(s) depicted in the surveillance films. | |
| Location, Time, and Date Examinations of photographic evidence can determine the location, time, and date that an image was taken. | |
| Authenticity and Image Manipulation Detection Photographic evidence including film, video, and digital images can be examined to determine whether the image is the result of a composite, an alteration, or a copy. | |
| Source and Age Photographic products including film and prints can be dated, and the source can be established by examining manufacturing characteristics. This can establish the time frame during which a photograph was taken. | |
| Cameras Cameras seized as evidence can be examined to determine whether a specific camera exposed a specific image. Digital cameras, including digital-video cameras, can be compared with digital images and video clips to determine whether a specific camera captured a specific image or video clip. | |
| Videos Black-and-white and color photographic images can be produced from video images for enlargement and used in courtroom presentations. | |
| Automobile Make and Model Identification Vehicles depicted in surveillance images can be compared with the National Automotive Image File to determine make and model. | |
| Child po*nography Examinations The seized images of child po*nography can be compared against images in the Child Exploitation and Obscenity Reference File to identify the source of the images. Video clips can be examined to determine if any of the people and scenes depicted in the video clips are also recorded as still images in the File. Video clips and still images can also be examined to determine if they depict recordings or images of real people and events or whether they represent computer-generated subjects and events. Questions concerning image analysis examinations should be directed to 703-632-6222. Questions concerning image analysis evidence should be directed to 703-632-6191. Follow the Evidence Submission directions including Requesting Evidence Examinations and Packaging and Shipping Evidence.
Submit original evidence (e.g., film or videotape) whenever possible because it contains the greatest level of detail. If the originals are unavailable, submit first generation photographic prints or videotapes. Process all film, including bank surveillance film, prior to submitting. When requesting forensic examinations based on video images, queue the original videotape to the approximate time of the pertinent area. State in a communication the date and time of the pertinent area and use the date-time stamp on the images or the counter indicator (set from the beginning of the tape at 000). If prints from the relevant frames are available, submit them for reference. Arrest or known photographs of suspect(s) for comparison with questioned images must depict the suspect(s) from many angles similar to the questioned images. If a facial comparison is requested, ensure that the suspect(s)' face or head fills more than half of the frame. If questioned images show tattoos or marks, include photographs of the same areas on the known suspect(s)' body. When taking known photographs for comparison with questioned images, use 35mm black-and-white film. If color film is used, include a color chart in the photographs. Do not mark or cut items submitted for comparison (e.g., clothing or firearms) where they are visible in the questioned images. Physical items such as clothing and firearms must first be submitted to the Laboratory for other examinations before they are submitted for image comparison. If photogrammetry is requested, include the dimensions of the scene to the nearest 1/8 inch and include a diagram or print from the surveillance film indicating the location of the measurements. Include one diagram or print for every angle used in the scene. Do not touch or move surveillance cameras except to remove the film. Submissions for comparison with the Child Exploitation and Obscenity Reference File must be limited to no more than 30 images. Call 703-643-6222 for specific instructions. | |
| Ink Examinations Examining inked writing in conjunction with other techniques (e.g., handwriting analysis, watermark identification) can provide details regarding document preparation. The composition of writing inks varies with the type of writing instrument (e.g., ballpoint pen, fountain pen, porous-tip pen) and the date of the ink manufacture. In general, inks are composed of dyes in solvents and other materials that impart selected characteristics. Ink analysis is usually limited to comparisons of the organic dye components. When ink formulations are the same, it is not possible to determine whether the ink originated from the same source to the exclusion of others. Examinations cannot determine how long ink has been on a document. Questions concerning ink evidence should be directed to 703-632-8441. Follow the Evidence Submission directions including Requesting Evidence Examinations and Packaging and Shipping Evidence.
| |
I'm not going to go into the basics of photography here. There are other pages on the web that will do it much better than I can. Actually, this section is going to be pretty short. There are just a few particulars to hit on, and a couple of interesting techniques. The first thing that needs to be done after securing the crime scene is photographing it. This creates a permanent record of the condition of the crime scene, one that is incontestable. First, take a picture that shows where the scene is; a shot with a street sign with the crime scene location in the background. Take pictures of the areas around the crime scene; alleys, dumpsters, rear areas, neighboring structures and even the structures across the street. Next, take pictures of the outside of the structure, showing points of entry and exit. Enter the structure, taking shots that show the locations and layout of the rooms. Take pictures of the whole room where the crime took place. Take close-ups of the scene or body. All pictures of items of evidence, which will be covered in the next paragraph, should be take both with and without a scale (a small ruler showing the size of the object). Take pictures with the scale to show the size of an object. Take pictures without the scale in case its presense in the picture blocks other evidence. What items are photographed at a crime scene? Bullet casings; photograph as a group and photograph individually. Photograph any dropped items, foot prints or animal tracks. If a homicide, photograph the body or bodies. Photograph any toolmarks, bitemarks or skin impressions. Basically, anything that might be evidence is photographed. Imprint evidence requires extra measures. Shoe imprints are photographed individually and as a series or group. Shoe imprints need to be lit from the side to show as much detail in the imprint as possible. Tire imprints are photographed from above as a whole. If the tire imprint is four feet long, then a picture showing all four feet is taken. Detail pictures are then taken showing one foot sections, each picture overlapping the one before it. This way, specific detail can be show and the overlapping pictures lined up to show the whole print. Again, all pictures are take with and without a scale. There is a special technique for no light situations. This technique is useful outdoors at night (perhaps a car accident scene), or in situations where the room is too big to light or there is no light available for pictures to be taken (such as a burnt out warehouse arson). The camera is set on a tripod with its shutter locked open. The photographer walks to several points in the room, popping off the flash, which is held in his or her hand. Each time the flash goes off, the film in the camera is exposed to another part of the room. The photographer does not appear as he/she is behind the flash and does not get exposed to the light when it pops off and only moves around the room while it is still dark. Remember, the film in a camera captures light. If there is no light, you can walk around in front of a camera all you want and never show up on the film. Video is also used to film crime scenes, taking long sweeping shots that take in everything in an unbroken time frame. The problem with video is, camcorder microphones will pick up the officers talking in the background, which can sometimes be embarrassing when the tape is replayed in court. |
|
|
|
|
< scrollAmount=9 scrollDelay=146 width=392 height=19 align="middle" border="0">copyright by H. v.d. Nieuwendijk, suggestions can be mailed to: ---> ---></> e--mail |
|
|
|
Most prints are not useful for a comparison, there is not enough information available in the print. Prints can be to old, to small, to dirty or damaged. Most fingerprints disappear after some days or some weeks. To know how to look at a print I am going to tell you the basics of fingerprints. . The right definition of a fingerprint is strictly speaking the print (stamp) that a finger left on an object. Besides fingerprints there are also other parts of the body that can leave good prints:
To understand where we are talking about we have to take a good look at the tips of our fingers. Use a magnifying glass to see it better! mailto:dacty@xs4all.nl Her you see lines in the skin that are higher than the surface. The lines are going in a certain direction. They look like a small dike with a dry ditch in between. This lines we call friction ridges. Sometimes there are small islands in between. The friction ridges are not always the same length, and are not always going in the same direction. Sometimes a line stops or splits. That are the unique points. mailto:dacty@xs4all.nl And that is the clue for the great mystery of the fingerprint! Because there are so many lines on a fingertip and because they are stopping, starting or slitting so many times there are no two persons in the world that have the same fingerprints. Even a square millimeter is different at all persons! 2. How does a finger leave a mark on an object?
3. Why are fingerprints important? Searching with fingerprints has only one big reason: To determinate to whom the print belongs. An identification of an fingerprints is for 100% reliable. There is no other technique that can establish such a high standard. Even DNA does not give absolute certainty. A twin does have the same DNA structure but does have totally different fingerprints. mailto:dacty@xs4all.nl The fact that everyone in the world has different fingerprints can be used in very different ways.
All around the world there are different standards to make an identification. In some countries it will be called an identification if there are somewhere between 7 to 15 identical points in the mark and fingertip. Other countries leave the identification over to the expert. It always is important that the are no unexplainable differences! We talk about explainable differences for instance when there is a scar on the skin, dirt in the background or on the finger, warts or differences because of the flexibility of the fingertip. 4. How do we search with a mark? To compare a mark it is necessary to have the prints of a person. To take the fingerprints of a person most of the time ink is used. Some ink is put on the fingertip and than the fingertip is pushed on a piece of paper. The fingerprinting is mostly done at the police. They take prints of fingertips and of the palm of the hand. In some cases prints can be made from the foot. To take someone's prints on free will can be done at persons that want to prove they are innocent or on identity papers. Sometimes prints are taken from unknown or dead people. 5. How can fingerprints made visible? There are a lot of techniques that make it possible to make invisible prints visible. And to store them for further investigation. The mostly used method is with powders. The powder will stick on the fat that has left there by the fingertip. A method that is used very much is working with superglue. A little bit of superglue is being heated and it will vaporize. The vapor attaches to the fingerprint and that will become visible. Do not try this at home because it is toxic and it must be done in a laboratory. To find prints on paper you can use a fine iron powder. The iron will stick on the fingerprint. There are a lot of other methods with all kinds of light or chemicals. 6. The way fingerprints makes figures. Global there are 3 groundshapes: From the shapes above a lot of other figures can be made but these are the most important. 7. The structure of a fingerprint. https://www.gironet.nl/home/hansnwdk In a fingerprint there can be a lot of lines. But there can be a great different in the amount of lines between one person or the other. 8. The future of the fingerprint. And in the future there will be the need for a lot of people that can work with fingerprints. Because a computer can search for a matching print but it can never tell if it is a real identification. There is always the need for people that have to check the results of the computer. Hans van den Nieuwendijk, https://www.fingerprints.demon.nl/ E-mail mailto:fingerprints@fingerprints.demon.nl |
< scrollAmount=9 scrollDelay=146 width=392 height=19 align="middle" border="0">copyright by H. v.d. Nieuwendijk, suggestions can be mailed to: ---> ---></> e--mail |



































 Anupamaa
Anupamaa
 Tu Juliet Jatt Di
Tu Juliet Jatt Di
 Naagin 7
Naagin 7

